Something pretty incredible happened last week. A clinical trial for a new gene therapy for Huntington’s Disease – a devastating condition with no known cure – showed 75% slowing of disease progression. This is huge. I wanted to take this opportunity learn for myself and share how this new therapeutic works. Let’s go.
Contents:
- First, a bit about Huntington’s Disease
- The genetic basis for Huntington’s Disease
- New breakthrough gene therapeutic
First, a bit about Huntington’s Disease. This is a genetic condition that runs in families with an ‘autosomal dominant’ inheritance pattern, basically meaning that if you have the Huntington’s Disease gene, you will develop Huntington’s Disease later in life, and as a parent there is a 50% chance you’ll pass this on to your biological children too. It’s a devastating and life-altering diagnosis. The disease starts showing aged 30-50 and pretty much affects all facets of life including inability to control movements, altered reasoning/ cognition, and worsening mental health. It gradually progresses until it is debilitating, and then fatal. There is currently no cure, and no treatments that can slow it down.

This brings up all kinds of ethical issues. Should people who have parents with Huntington’s Disease get genetically tested? This can be a great source of anxiety and guilt.
- Some people understandably don’t want to be tested and find out with certainty that they have a 100% chance of developing an incurable and progressive condition in their middle age. If they aren’t tested, they are still aware there is a 50% chance they are affected. People also worry about the implication on employment and insurance costs, etc. if they receive a positive test result – although technically there are laws that protect patients from being discriminated against for choosing to get tested.
- Others want to know, so they can factor this into their life planning (savings for their family, arrangements for care later on, etc.) and family planning (they may choose not to have biological children if there is a 50% chance of passing this on).
There is no right answer, and it is heartbreaking either way for patients and their families.
So what is the genetic basis for Huntington’s Disease?
First, a quick recap. We spoke before about the link between DNA base pairs, genes, and proteins. DNA bases are the building blocks of genes. A defined segment of DNA is a genes. Each gene codes for a protein – and when the gene (defined DNA segment) is expressed using mRNA via the processes of transcription and translation, the protein is produced. Proteins can serve various functions in the body – for example, enzymes, antibodies, and neurotransmitters are all proteins. There are also ‘non-coding’ parts of DNA called ‘introns’ that don’t encode any proteins themselves, but actually serve to regulate gene expression (i.e.: increase or decrease the number of proteins produced from the target gene).
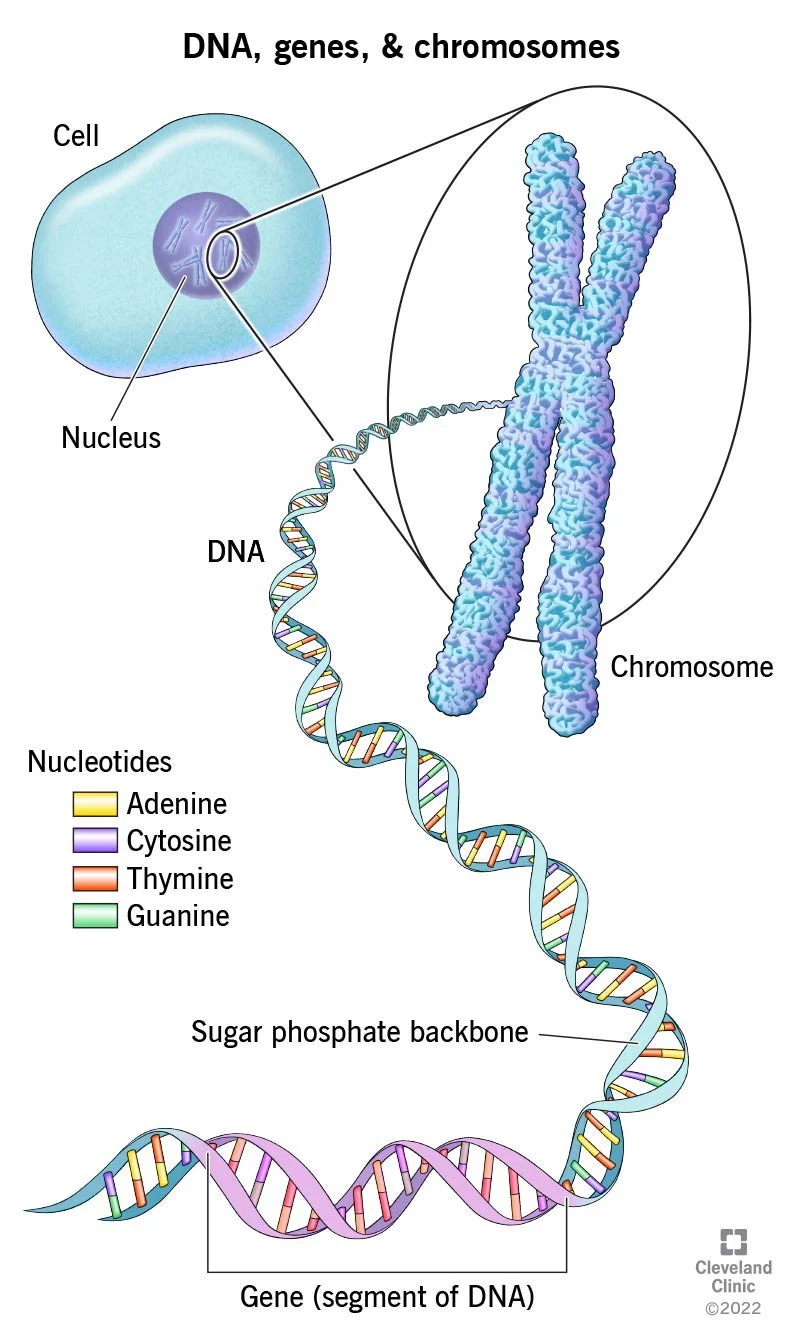
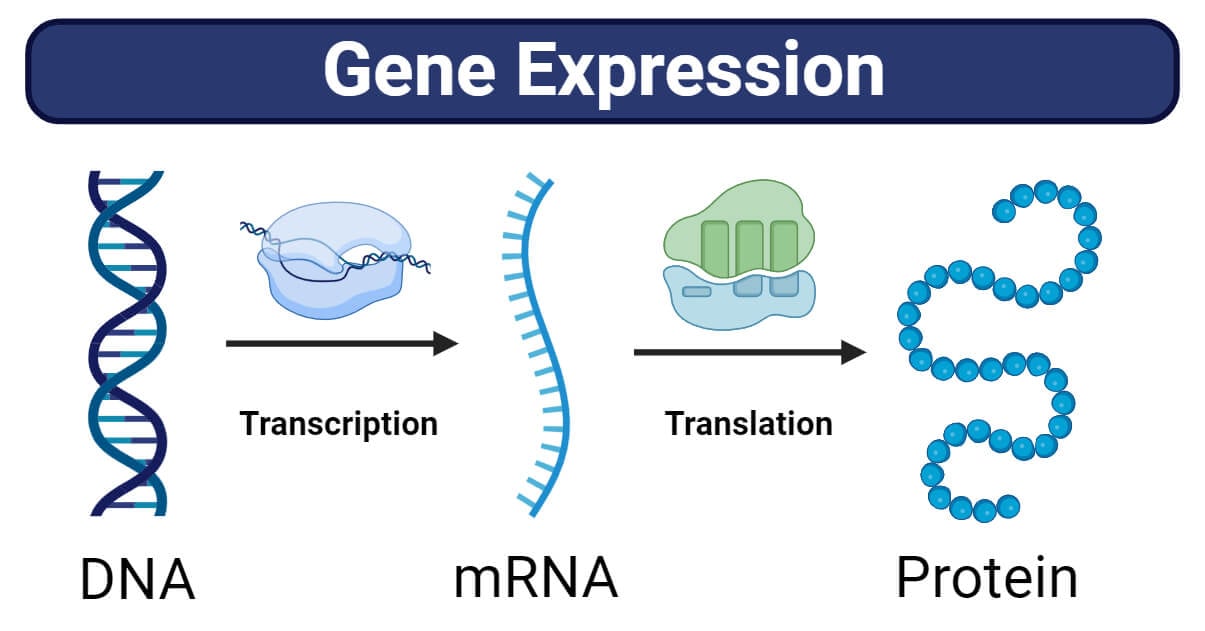
So if there is a mutation/ abnormality in the base pairs, this maps to a gene mutation, which then leads to problems in the synthesis and function of the protein that that gene encodes. If that protein doesn’t work, or works abnormally, it can cause disease.
As mentioned before, Huntington’s Disease is an autosomal dominant condition which means every individual with the mutated (diseased) gene has a 100% chance of developing the disease, and a 50% chance of passing on the mutated gene to their children. Huntington’s Disease is caused by an abnormally high number of ‘CAG’ base pairs in the DNA which leads to a mutation in the huntingtin gene (HTT), which then leads to a faulty HTT/ huntingtin protein. This protein is essential for neurological function, and the mutation leads to the protein becoming toxic and damaging neurons in a part of the brain called the basal ganglia leading to involuntary movements, cognitive decline, and psychiatric problems.

Generally speaking, the more CAG repeats, the higher the disease penetrance, and the higher the likelihood of earlier and more severe disease (there is more nuance to this but we can leave that aside for now).

Now let’s learn about the recent breakthrough gene therapy for Huntington’s Disease.
- This is based on early results from a Phase I/II clinical trial involving 29 patients.
- The drug is called ‘AMT-130’ and is made by uniQure, an Amsterdam-based company.
- The goal of this treatment is to permanently block production of the toxic mutated Huntingtin protein with a single dose of the drug into the targeted cells.
- A harmless virus (‘AAV’) is used to deliver an microRNA (miRNA) strand – a small intron strand that regulates gene expression – directly into the affected neurological cells in the basal ganglia. This therapeutic miRNA strand encodes a silencing protein that blocks the target cell from producing the faulty Huntingtin protein responsible for Huntington’s Disease.
- The therapeutic is infused slowly and delivered precisely using real-time MRI scanning to guide a small tube through small holes in the skull into target regions in the basal ganglia. This is no small procedure, and requires 12 – 18 hours of complex neurosurgery.
- Once the virus with the therapeutic strand is delivered to these target regions, it continues to use the body’s own gene expression mechanisms to produce the silencing protein and block production of the faulty Huntington protein indefinitely. So the therapy becomes self-sustaining in itself.
Isn’t that cool!?
Here’s a [simplistic] diagram from the BBC that serves as a good visual.
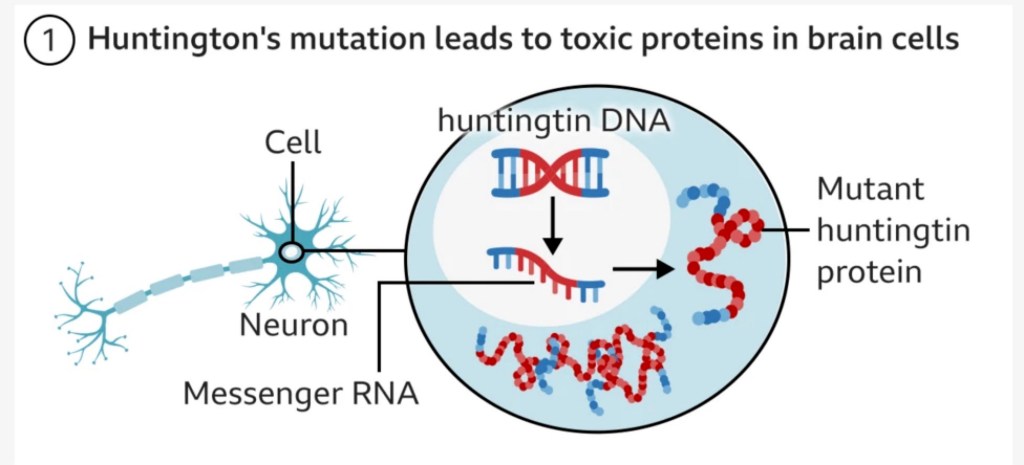

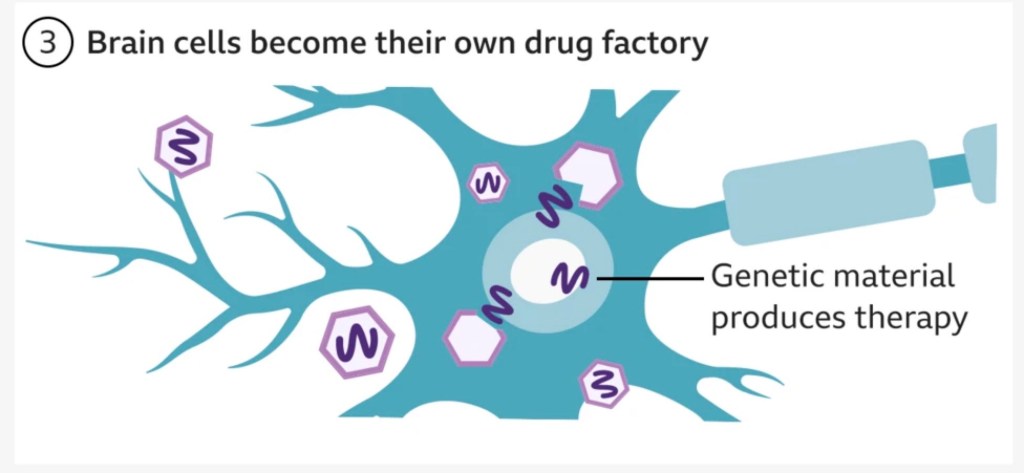
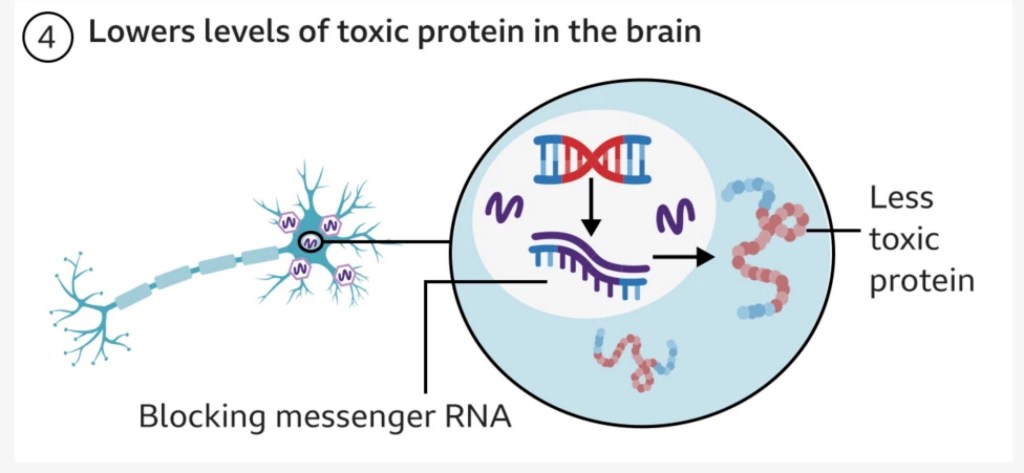
The good news is that early results show a statistically significant 75% slowing in disease progression three years post-surgery, meaning that patients on the drug saw decline in their symptoms over four years rather than one, which in the longer term could lead to decades of a better quality of life. People seemed to tolerate the procedure well, with only minor side effects such as headaches. One patient who was medically retired was able to return to work, and others in the trial are still walking despite being expected to need a wheelchair. uniQure plans to seek regulatory approval for the drug as soon as next year. For a disease without a cure and no treatments, this is life-changing for patients and their loved ones. I can’t stress what a beacon of hope this is for those heading towards an inevitable debilitating disease.
The bad news is this is likely to be an extremely expensive therapeutic at an estimated cost of > $1m per person, which may make the treatment unaffordable for many patients. It also requires the expertise and facilities for complex neurosurgery – which makes it a difficult treatment to deliver globally and could lead to selective access and widen health inequities. It’s also important to note that the trial shows the drug slows disease, but does not cure it.
The best part about these drugs is that they are TARGETED to the affected cells (which theoretically limits any complications from gene therapy elsewhere in the body), and that they continue to have a lasting effect on suppressing disease. There is hope that treating patients with the mutated gene before they develop the disease could stop the disease from presenting altogether – a prevention trial to assess this would be a good next step.
I really enjoyed diving into this, and I am crossing all fingers and toes that this revolutionary therapeutic comes through in Phase II/ III trials. This would be a blueprint for gene therapies for previously incurable diseases. There are similar studies being done for AAV viral vector gene therapies for Parkinson’s Disease too. It’s an exciting time to be in gene therapy.
PS: Apparently there is a rival gene therapy for Huntington’s Disease by Spark Therapeutics (Roche) that also uses the AAV virus to deliver a similar therapeutic molecule. It entered clinical testing earlier this year but results are pending. More crossed fingers and toes.
Sources:
https://www.ucl.ac.uk/news/2025/sep/gene-therapy-appears-slow-huntingtons-disease-progression
https://www.bbc.co.uk/news/articles/cevz13xkxpro

Leave a comment